EU Clinical Trials Regulation 536 2014 (CTR) has replaced Clinical Trials Directive 2001/20/EC (EU-CTD) as of January 2022. Regulation EU No 536 2014 is applicable to national and multinational clinical trials in the EU. The Clinical Trial Regulation was implemented to simplify and accelerate clinical trials within the EU, so new treatments would become faster available to patients. Regulation 536 2014 assures that EU member states remain an attractive location to conduct clinical trials.
Regulation (EU) No 536/2014 timeline (infographic)
EU clinical trial regulation 536/2014 for drug developers
This blog will go into detail about the EU clinical trials regulation 536/2014 and its implications for clinical trial sponsors. After reading, you will understand the basics of the regulatory clinical research requirements. More importantly, you will be able to estimate the timelines for your clinical trial application and know how to submit your study for approval.
If you need help with meeting the regulatory requirements for clinical trials, please contact us. Our certified team will be happy to assist you.
Regulation (EU) No 536/2014 questions & answers
Clinical research regulations often lead to questions and the clinical trials regulation EU No 536 2014 CTR is no exception. As a clinical research organization (CRO) we at TRACER often get questions from our sponsors about applicable regulations. Many of those are about EU CTR 536 2014. We’ve listed the most common questions & answers for this topic in this blog. If your question is not listed, or if you would like to receive more information, feel free to send us an e-mail at info@tracercro.com.
What is EU Regulation 536 2014?
What is the EU regulation No 536 2014? It is also referred to as the European Clinical Trial Regulation (ECTR) or EU Clinical Trials Regulation (EU CTR) and is currently the latest version of the European legislation on clinical research on medicinal products and trials in the European Union. Its implementation has significantly changed the process for starting clinical trials in Europe for everyone who is involved in this type of research. All clinical trials must be submitted according to CTR.
The EU CTR includes the medical ethics review process, safety reporting, and the publication of trial data.
What is the main difference between regulation 536 2014 and clinical trial Directive 2001 20 EC?
EU 536 2014 differs from EU Directive 2001 20 EC. The main difference is that clinical trial legislation has become a regulation, rather than a directive. Namely, EU-CTD did not carry the same weight as a regulation. Every country implemented its own legal requirements through national legislation. This means that previous conduct of clinical trials had to be approved in every country separately. This made the approval and execution of multinational clinical trial applications complex. It also decreased the availability of clinical trial information to the public. That is why with EU CTR identical rules need to be followed across all EU member states.
How long is the transition period for EU CTR?
Regulation (EU) No 536 2014 will replace national law, with a transition period of three years. This means the EU CTR transition period ends on January 31st, 2025.
Up until January 31st, 2023, you could still submit your clinical trial application under any system, regulation 536 2014, or 2001/20/EC. From now onward, you are only allowed to submit clinical studies according to the EU-CTR. If your study was approved under the directive before January 31, 2023, it can still be regulated according to the directive until 31st of January 2025. The latter also marks the end of the transition period, which means all clinical trials must be regulated under the EU-CTR.
Do I submit my clinical trial according to regulation 536 2014 or Directive 2001/20/EC?
As of January 31st, 2023, all new clinical trial applications need to be submitted according to the EU clinical trial regulation.
Do clinical trial applications change with 536 2014?
The type of information you need for your clinical trial application (CTA) remains the same. However, because of the new clinical trial regulation, how you submit your study differs significantly from before. Many different templates are used. You can find the link to the templates under this article in the download section. Under the EU CTR regulation, sponsors can submit a single application for a clinical trial. This application is valid in all EU member states involved in the trial, replacing the need to submit separate applications to each country. This single application is submitted via an online system called the Clinical Trial Information System (CTIS).
What is the Clinical Trial Information System (CTIS)?
The Clinical Trial Information System (CTIS) is an electronic portal introduced by the European Medicines Agency (EMA) as a part of the new EU regulation (536/2014). The goal of CTIS is to streamline clinical trial submission and information provision.
Before EU-CTR, a clinical trial application (CTA) was submitted to local regulatory authorities and ethics committees of each EU member state. They had to separately provide regulatory approval for the clinical trial. With the clinical trial information system sponsors can submit a single electronic CTA for all countries in the European Economic Area (EEA). This single point of reference and the very strict timelines accelerate the regulatory approval process and improve trial information management.
How does CTIS improve transparency?
CTIS focuses on improving clinical trial transparency. Through the EU Clinical Trials Database, the system makes crucial information about clinical trials publicly accessible. The contents are decided by sponsor, type of study, and phase. This includes clinical trial design, objectives, and results. It also automatically updates trial-related information, reducing administrative tasks for sponsors and authorities.
Beyond administrative benefits, CTIS also contributes to patient safety and pharmacovigilance. It supports the reporting and tracking of safety data and serious adverse events during the course of clinical trials.
How do I submit a clinical trial application in CTIS?
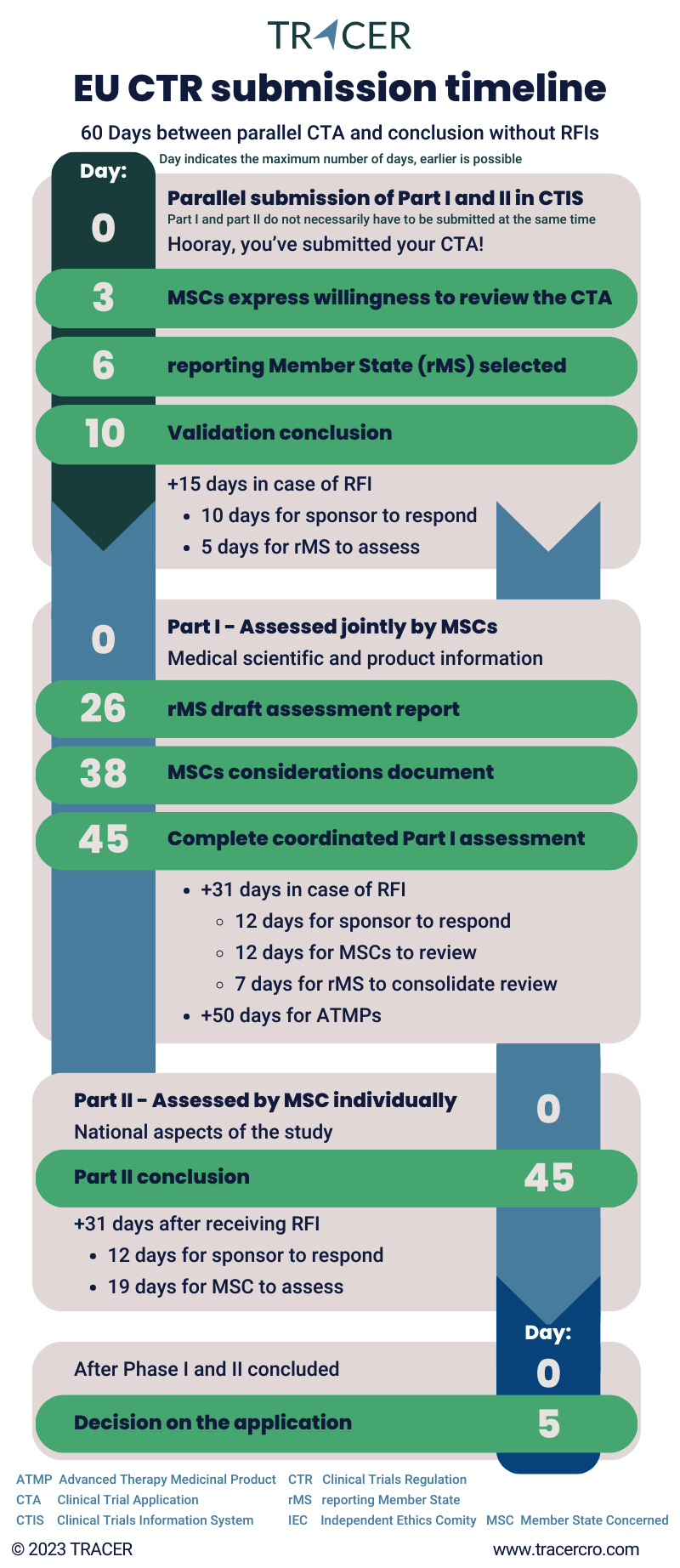
The EU CTR submission process, according to regulation EU No 536 2014, is divided into several stages: validation, part I assessment, part II assessment, and decision on the application. The following paragraphs will provide information on each stage.
Validation of (initial) application by member states
After submitting an (initial) clinical trial application, the member state authorities have 10 days to validate it. They consider whether the trial falls under the clinical trial regulation and if the application dossier is complete. If this timeframe is exceeded, the trial gets validated automatically. In case they send you a request for information (RFI), up to 15 days can be added to the validation process. Tip: Check CTIS daily for RFIs and always answer an RFI in time to prevent your application from lapsing.
Usually, by day three of the validation process all involved Member States will express their willingness to continue reviewing the clinical trial. For multinational clinical trials, one of the member states acts as the reporting member state (rMS). This country is usually selected by day six.
Which member state becomes the rMS?
Sponsors have the option to indicate their preferred rMS. However, which member state becomes the rMS depends on the availability and the number of times that a Member State has been the rMS in the past. The role of rMS is distributed evenly among each Member State.
Who is the rMS for national clinical trials?
For a national clinical trial, the reporting Member State (rMS) is the Member State (MS) to which the clinical trial application is submitted. This is the country where your trial will be conducted.
What does a rMs do?
The reporting Member State supervises the assessment process and draws up the assessment report in coordination with the other Concerned Member States (CMS).
Can I choose the reporting member state (rMS)?
When you submit your clinical trial application, you indicate which countries are involved in your multinational clinical trial. You also have the option to specify which member state of the ones included in your study, you prefer to be the rMS. For example, if you include the Netherlands, France, and Germany, it can be smart to let the Dutch authorities review your CTA due to its beneficial regulatory environment. Read our blog about reasons to conduct your clinical trial in the Netherlands.
Please note that the member state that you select to review your application does have the right to decline its review role.
Main benefits of conducting a clinical trial in the Netherlands
As a Contract Research Organization in the Netherlands, we at TRACER, are experienced with getting clinical trials approved here. There are some major benefits our sponsors experience by conducting their study in The Netherlands.
Good communication with the Medical Research Ethics Committee (MREC)
With EU regulation 536 2014, the clinical trial law is the same for all EU member states. However, how a Medical Research Ethics Committee (MREC) reviews an application can be different. For multi-country studies, the responsible member state will first look at the documents and other member states will look more at country-specific factors.
We’ve always experienced fast communications with METC (Medische Ethische Toetsingscommissie, the Dutch MREC), CCMO (Centrale Commissie Mensgebonden Onderzoek, Dutch Central Committee on Research Involving Human Subjects), the Medical Review Board (MRB) and other involved parties.
One approval applies to every clinical site
Approval from one hospital applies to any additional hospital you would want to include in your multi-center study. This accelerates your approval process, as you do not have to submit separate requests for every hospital, nor does the IRB of each hospital go through the evaluation process again. Exceptions for this procedure are clinical trials with minors, gene therapy, and vaccines.
Good cooperation between local hospitals
There is good cooperation between local hospitals; hospitals see each other more as colleagues than as competitors. Especially between academic hospitals, there is a scientific ecosystem with cross-communication between professionals working at different locations.
Good inclusion rates and lower fees
Many hospitals have a large service area, and therefore, are in contact with many patients. Also, Dutch patients display a high willingness to participate in clinical trials which can prevent inclusion problems.
At the same time, the Netherlands offers competitive per patient costs. In general, the price of clinical therapies and diagnostics are higher in for example the United States than in most other western countries.
Ask your questions about clinical trial regulations in the Netherlands
If you are considering conducting a clinical trial in the EU and you want to know more about clinical trials in the Netherlands, get in contact with TRACER. One of our clinical trial managers is happy to schedule a meeting to answer your questions and advise on specific Dutch clinical regulations regarding clinical studies.
Part I of the EU CTR application dossier (central)
Part I consists of the medical scientific and product information. It is jointly assessed by all member states. By day 26 the rMS should have distributed a draft assessment report to all other Member States. In the next 12 days, all member states document their considerations. The complete coordinated Part I assessment takes a maximum of 45 days.
Reasons why a CTR application may take longer
In case you are subject to a request for information (RFI), the process can take up to 31 days longer. This includes time for the sponsor to respond and the MS to review the answer. On certain medicines for human use clinical trials regulations can be different. If you are developing medicines based on genes, tissues, or cells, otherwise known as Advanced therapy medicinal products (ATMPs), Part I can be extended up to +50 days.
What documents do you need to submit for EU-CTR part I?
Documents that you need to submit for part I include:
- Cover letter with EU Trial Number
- EU Clinical Trial Application form (in CTIS)
- The final version of the research protocol and protocol synopsis
- Investigator’s Brochure (IB)
- Declaration of release by Qualified Person (QP)/permit for the production or import of investigational medicinal products
- Investigational Medicinal Product Dossier (IMPD) or Summary of Product Characteristics (SmPC)
- Label of the investigational medicinal product (IMP)
- Dossier for Auxiliary Medicinal Products (AxMPs) (if applicable)
- Data safety monitoring board (DSMB) Charter (if applicable)
- Scientific advice and Paediatric Investigational Plan (PIP) (if applicable)
- Description of the contents of the labelling of the investigational medicinal product
Part II of the EU CTR application dossier (national)
Part II covers the national aspects. It is assessed by each concerned Member State individually and like Part I it takes up to 45 days. Part II assessment can be done in parallel with Part I but can also be submitted subsequently/after Part I has been approved.
Part II of the EU CTR includes:
- Recruitment procedures, including advertisements and copies of materials
- Subject information, Informed Consent Form (ICF), and other subject information material, i.e., information leaflets
- List of planned clinical trial sites, investigators, and number of subjects
- Investigator’s CVs and qualification/training documentation
- Statement of suitability of clinical trial site facilities also known as Site Suitability Declaration
- Policy or proof of subject insurance or indemnification
- Brief description of the clinical trial’s financing
- Information about compensation paid to subjects and investigators
- Documents stating the compliance on the collection and use of personal data and the collection, use and storage of biological samples
General part of EU CTR form:
- Proof of payment of fees (if applicable)
- Declaration from the sponsor to process data in accordance with the General Data Protection Regulation (GDPR)
What is the difference between Part I and Part II EU CTR clinical trial application?
Where Part I includes general clinical trial information and is reviewed by all Member States, Part II of the application dossier includes locally applied documents and is assessed by each included Member State individually.
Decision on your clinical trial application
Once Part I and Part II of the clinical trial approval process under EU CTR 536 2014 are concluded, a decision on your clinical trial is made. The conclusion of Part I assessment needs to be supported by all Member States Concerned (MSCs). The decision is made by each involved Member State and communicated with the rMS. This takes a maximum of 5 days. The decision is then communicated by the sponsor’s rMS through the Clinical Trials Information System (CTIS) and a letter of approval.
How long does clinical trial approval EU CTR take?
EU member states have a maximum of 60 days to evaluate an initial clinical trial application according to regulation 536/2014. However, this time frame can be expanded depending on the number of RFIs. During your application, it is recommended to check CTIS daily because you don’t receive a notification of an RFI in any other place!
Clinical trial amendments
Once a clinical trial is approved a sponsor can submit an amendment through non-substantial or substantial modifications. If you want to make a non-substantial change, this does not require resubmission. Substantial ones do.
What is considered substantial modification clinical trials?
Under EU clinical trial regulation No 536 EC 2014, substantial modifications encompass changes affecting safety or data. Such changes require resubmission and assessment of relevant parts—Part I, Part II, or both.
For example, changes to the protocol related to the inclusion criteria or safety monitoring. Amendments related to the IMP can also be substantial modifications. For example, change of formulation or additional toxicology data. Finally, amendments related to trial arrangements such as changing the coordinating investigator also fall under this principle.
Substantial changes undergo a validation phase to ensure alignment with the correct application section. To include an additional country, they will assess Part II for that Member State. While Part II is obligatory, the Member State might provide Part I insights, triggering reassessment as necessary.
EMA specified timeline of CTIS/CTR tasks
The introduction of EU CTR comes paired with a stricter clinical trial application timeline. Including timely responses to RFIs.
The European Medicine Agency (EMA) indicates specific rules about due dates for CTR/CTIS tasks. Things you should know about EMA clinical trial regulation regarding dates:
- The time zone is Central European Time (CET)
- A task starts the first working day after it has been created
- Tasks can only start on a working day
- A task due date cannot fall on weekends or public holidays. If this happens, it will be moved to the next working day
- A minimum of two consecutive working days is given to complete a task
- The task timer is paused during the holiday period from December 23rd up until January 7th
EU CTR RFI deadline
As per EU CTR guidelines, responding to an RFI can take up to 12 days, though practical timeframes may be shorter. Meeting the RFI deadline can be tough, especially when additional steps are needed before CTIS submission. A correct initial document submission is crucial for on-time responses. At TRACER we value our good reputation at regulatory authorities and always ensure timely replies. If meeting deadlines is important to you, get in contact to find out how we ensure timely regulatory responses.
How long does a sponsor have to respond to an RFI?
Sponsors have 12 days to respond, starting the next working day from the moment they’ve received the request for information. Unless stated otherwise by the member state, this will not exceed 12 days. The MSCs can determine a shorter period available for sponsors to respond to the RFIs (i.e. maximum of 10 days for validation RFIs and a maximum of 12 days for assessment RFIs). However, a minimum of one working day must be given for sponsors to respond.
How do Requests for Information (RFI) influence timelines of your initial application?
Request for information impacts the timelines of your initial application. Once you receive an RFI, you have 12 days to respond. Not responding in time, will result in your application lapsing. After receiving your answer, the reporting Member State then has 12 days to evaluate the answers and another 7 days to consolidate the review.
Keep in mind that any substantial modification and additional Member State countries you’ve added to your submission can result also in longer timelines.
What happens if you do not respond to an RFI in time?
If you do not respond to an RFI within the limit of 12 days you risk a lapsed application. This means you will have to start your clinical trial application again. In that case, you receive a new EU CT number (the initial one ends with -00 and the new one with -01). All documents and info remain, but you have to change all document dates and resubmit, so you can go again through the validation phase. To avoid this, it is important to respond on time and monitor any incoming RFIs closely. Important: The CTIS does not notify you via email if you receive an RFI. You can see the notification in CTIS after you’ve logged in.
Does the EU CTR allow parallel submissions?
The CTIS system does not allow parallel submission of requests. This means that while you have any ongoing assessments, such as initial application, substantial modification, and/or request to include another EU member state, you cannot submit any other request for that trial. Be aware that this limitation can affect the timing of your clinical trial.
EU regulation 536/2014 safety reporting
Under the Clinical Trial Regulation 536 2014 all unexpected events, urgent safety measures, and annual safety reports should be reported in the CTIS. If your clinical trial was approved under Clinical Trials Directive 2001 20 EC of 4 April 2001 then information should be reported to the applicable local and EU authorities.
Clinical Trial oversight
The Clinical Trial Information System (CTIS) simplifies communication between sponsors and regulators, aiding prompt safety updates. It also is a tool for ethics committees and regulatory authorities to collaborate, providing an added layer of safety oversight throughout the trial.
Sponsors are required to create risk assessment and risk management plans for each trial, identifying and managing potential risks. Additionally, safety reporting goes beyond trial completion. Sponsors are obligated to share summary results and safety reports with authorities.
Reporting of Adverse Events (AEs) and Serious Adverse Events (SAEs)
Adverse drug reactions cover all untoward and unintended responses to an Investigational Medicinal Product (IMP) related to any dose administered. However, Adverse Events (AEs) cover untoward medical occurrences in a patient or clinical trial subject without necessarily a causal relationship with any treatment. Serious Adverse Events (SAEs) involve more serious outcomes like hospitalization or disability. AEs and SAEs are therefore not necessarily related to IMP. When an AE occurs, relatedness to IMP should be evaluated. There are strict regulatory timelines to report AEs and SAEs, ensuring swift responses and participant safety.
Reporting of Suspected Unexpected Serious Adverse Reaction (SUSAR)
An SAE that occurs during research with a medicinal product may be a SAR or a SUSAR. SAR is the abbreviation for Serious Adverse Reaction, and SUSAR for Suspected Unexpected Serious Adverse Reaction. SAEs are reported in the annual safety report, SUSARs have to be reported in max 7 or 15 days to EudraVigilance. This applies for both EU CTR and the Directive 2001 20.
Downloads
Download all CTR templates here:
https://english.ccmo.nl/investigators/clinical-trials-with-medicinal-products-ctr/preparation-ctr.
In the Netherlands, the relevant sections for application and submission to the CTIS can be found here in English: https://english.ccmo.nl/investigators/clinical-trials-with-medicinal-products-ctr. On that page, you will also find the link to the clinical trial regulation pdf. The suppletion, regulation 2017/1569 on GFP for investigation medicinal products can be found here: https://eur-lex.europa.eu/eli/reg_del/2017/1569/.
CTIS handbook
The EMA provides an online handbook for the CTIS in PDF: https://www.ema.europa.eu/en/documents/other/clinical-trial-information-system-ctis-sponsor-handbook_en.pdf.
RFI FAQ
Download the RFI FAQ from EMA here: https://www.ema.europa.eu/en/documents/other/faqs-how-respond-requests-information-received-during-evaluation-clinical-trial-application-ctis_en.pdf.
Clinical Directive 2001 20 EC
Clinical Directive 2001 20 EC of 4 April 2001 has been repealed. However, it will still apply until 30 January 2025 for trial applications submitted before 31 January 2023 and submissions between 31 January 2022 and 30 January 2023 where Directive 2001 20 EC was opted. If this applies to you, you can find more information here: https://health.ec.europa.eu/medicinal-products/clinical-trials/clinical-trials-directive-200120ec_en. For new applications, read regulation EU 536 2014.
Used abbreviations
| AE | Adverse Event |
| ATMP | Advanced Therapy Medicinal Product |
| AxMP | Auxiliary Medicinal Product |
| CCMO | Centrale Commissie Mensgebonden Onderzoek (Central Committee on Research Involving Human Subjects) |
| CTA | Clinical Trial Application |
| CTIS | Clinical Trials Information System |
| CTR | Clinical Trial Regulation |
| DSMB | Data safety monitoring board |
| ECTR | European Clinical Trial Regulation |
| EEA | European Economic Area |
| EMA | European Medicines Agency |
| EU CTR | EU Clinical Trials Regulation |
| GDPR | General Data Protection Regulation |
| IB | Investigator’s Brochure |
| ICF | Informed Consent Form |
| IMP | Investigational Medicinal Product |
| IMPD | Investigational Medicinal Product Dossier |
| METC | Medische Ethische Toetsingscommissie (Dutch) |
| MRB | Medical Review Board |
| MREC | Medical Research Ethics Committee |
| MS | Member State |
| MSC | Member State Concerned |
| PIP | Paediatric Investigation Plan |
| QP | Qualified Person |
| RFI | Request For Information |
| rMS | reporting Member State |
| SAE | Serious Adverse Event |
| SAR | Serious Adverse Reaction |
| SmPC | Summary of Product Characteristics |
| SUSAR | Suspected Unexpected Serious Adverse Reaction |