Phase 0 exploratory studies have been around for more than 20 years, but many drug developers are still surprised when they learn about the possibilities. As an expert Phase 0 Contract Research Organization (CRO), we’ve received many questions over the years. This article bundles all of the answers. Most importantly to know, Phase 0 evaluates the biodistribution and pharmacokinetics of new drugs in a first-in-patient study. Phase 0 is conducted before Phase 1 and is therefore also referred to as ‘early Phase 1’. But how can a patient study be conducted without data on safety from healthy volunteers? This and other questions are answered below.
No time to read, or can’t find your question?
Ask your question directly
1. Under what names can Phase 0 studies be found?
To kick things off, let’s clarify the definition. When searching for early studies in humans, it is important to know exactly what to look for. This type of exploratory clinical research is referred to by various names. However, the contents of the research may vary, and some common names for Phase 0 are also used for other types of studies.
A good example is the term “exploratory trials”, used by the U.S. Food and Drug Administration (FDA) [1] to indicate Phase 0; however, in ICH guideline E8 (General considerations for clinical studies) [2], “exploratory trials” indicates the difference between Phase 2 (exploratory) and Phase 3 (confirmatory) studies. So, when searching for Phase 0 studies, check also the other criteria that are discussed later in this article.
ICH stands for The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use.
Commonly used names for exploratory Phase 0 research:
- Phase 0
- Exploratory trials and exploratory studies
- Early Phase 1
- Early phase exploratory trials
- Microdose study, also abbreviated to MD study or exploratory MD clinical study
- Exploratory IND study (or abbreviated, eIND study), based on FDA terminology
- Microtracer study (this name may be used when the drug is labeled with a tracer and administered at a microdose, but also when a microdose of a tracer is used combined with a therapeutic dose in a Phase 1 trial)
- Pilot study
In this article, we will mainly use the term Phase 0, as it clearly indicates the stage of the research at which it is taking place.
IND stands for Investigational New Drug
2. When can a Phase 0 study be conducted?
Drug developers can conduct Phase 0 studies while still in the preclinical phase, between single-dose toxicity studies in rodents and studies in large animals. This means that Phase 0 can be conducted with a minimum of safety studies in animals and without safety studies in healthy volunteers. Phase 0 uses low doses that are not intended to have a therapeutic effect, hence the low safety requirements. The required toxicological testing depends on the dose and dosing schedule of the Phase 0 trial, as stated in ICH M3 R2 [3].
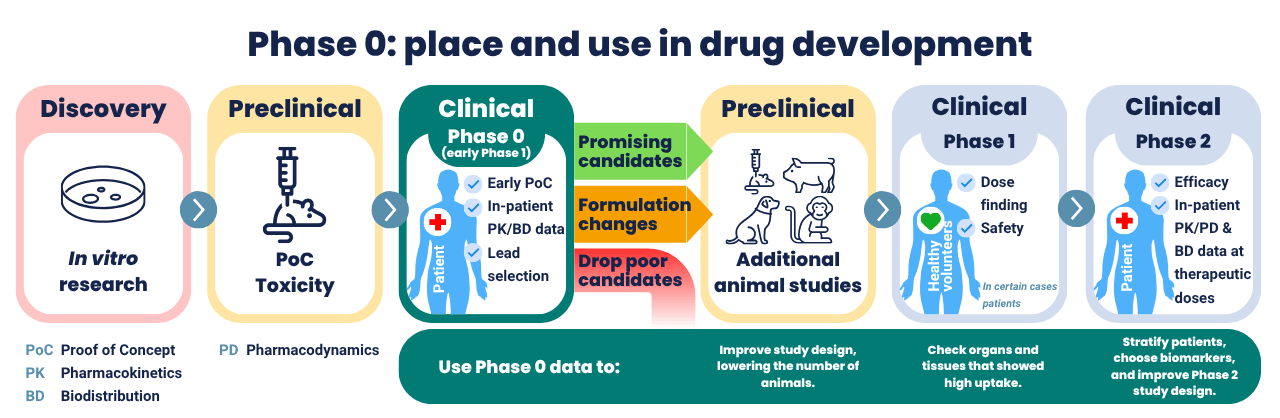
Principle of Phase 0
It basically boils down to this: you conduct the minimum preclinical research to show proof-of-concept and assess toxicity, and then you can test your drug at low levels in humans. With this data, you can make a go/no-go decision for further development of the drug candidate. The data obtained can be used to set up subsequent studies. They can be used to evaluate research in animals and animal models compared to patients and to observe potential dissimilarities between patients and healthy volunteers. More study goals will be discussed later in this article.
3. Is Phase 0 always a microdose study?
No, while Phase 0 is commonly conducted with a single microdose, you are allowed to evaluate the drug at higher doses. The ICH provides five approaches as examples in their ICH M3 guidance. There are multiple options for dosing and dose regimens in Phase 0. The most common option is a single microdose, but it’s not the only option. The second is a repeated microdose (up to five). The other three suggestions are non-microdose exploratory trials, but all are subtherapeutic.
4. What non-microdose and repeated dosing regimen is possible?
Here we list the other, non-microdose options as suggested by M3 R2 guidance.
Non-microdose study with a single administration
Starting at a subtherapeutic dose or, with regard to toxicity and Pharmacologically Active Dose (PAD) findings, at the anticipated therapeutic range. Each participant receives a single dose. The maximum dose is, with toxicity observed in animals and considered monitorable and reversible in humans, less than ½ of the No Observed Adverse Effect Level (NOAEL) in more sensitive species [4].
Non-microdose study with multiple administrations
Option 1: Starting at 1/50th of Area Under Curve (AUC) at the NOAEL in the most sensitive species. Dose regimen is limited to <14 days. The maximum dose is, without toxicity in both species, 1/10th of the AUC and with toxicity in one species, less than the NOAEL of the species showing toxicity, or ½ of the AUC of the species not showing toxicity (whichever is lowest). With toxicity in both species, follow regional guidance.
Option 2: 1/50th of the NOAEL in the most sensitive species on mg/m2 basis. Dose regimen <14 days and less than the duration of dosing in non-rodent. The maximum dose is in the anticipated therapeutic dose range, less than AUC NOAEL in non-rodent or ½ AUC NOAEL in rodent (whichever is lowest).
Keep in mind that these are merely suggestions by the ICH. Other study designs may be suitable. Furthermore, the higher the dose, the more preclinical data is required. All can be found in our exploratory trial approaches table.
Download exploratory trial approaches table

5. How much is a microdose?
A microdose is defined as a limit to the total dose of 100 microgram (μg). The total dose may also not exceed 1% of the NOAEL or 1% of the PAD (scaled on mg/kg for intravenous (i.v.) and mg/m2 for oral). The lowest of the limits mentioned should be used as the limit. For protein products, the limit is ≤30 nanomole (nmol). Read more about the dose and learn all aspects of this in-human study on our microdose study page.
6. What administration methods are available in Phase 0 studies?
Mostly, i.v. is used since this offers a 100% bioavailability (BA) of the already low microdose. However, other administration routes are possible. For example, oral, intranasal/inhalation, and topical administration. The route of administration influences the required toxicological package. What administration method to choose depends on the type of compound, the indication, and the research question. When using oral administration, keep in mind that BA is always lower, and BA from animal studies is often very different from that in humans.
Multiple cohorts with different administration methods
You may design your clinical trial with different cohorts for different types of administration. For example, to compare i.v. with oral administration for Absorption, Distribution, Metabolism, and Excretion (ADME). I.V. administration may be allowed, even if the drug is intended for oral administration.
7. What analytical measurement methods are used in Phase 0?
Sensitive methods to obtain data from a low dose are needed. Most common are Positron Emission Tomography (PET), Single Photon Emission Computed Tomography (SPECT), Accelerator Mass Spectrometry (AMS) using 14C-labeled compounds, and Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS) using non-radiolabeled compounds [5, 6, 7, 8, 9, 10]. We at TRACER often choose to label the drug with a radiotracer for PET or SPECT imaging. This method allows to obtain visual data and spatial information on whole body biodistribution (BD), pharmacokinetics (PK), and on- and off-target distribution in organs and tissues. Data from nuclear imaging can be quantified to calculate tissue concentrations of the radiolabeled drug. When quantitative analysis is performed at several timepoints after injection of the radiolabeled drug, this data can be used for dosimetry calculations.
Limitations of AMS and LC-MS/MS
Both AMS and LC-MS/MS are used in clinical studies to study drug concentration, composition, metabolites, etc., but don’t deliver imaging data and spatial information like PET or SPECT. AMS and LC-MS/MS are limited to ex vivo analysis of bodily fluids like plasma, serum, urine, feces, cerebrospinal fluid, etc., and in biopsies. Of the two, AMS is more sensitive than LC-MS/MS, but requires 14C labeling. A combination of methods can be used, for example, AMS and LC-MS/MS [11], AMS and PET [12, 13, 14], or, very commonly, PET or SPECT accompanied by MRI or CT to add an anatomic data layer.
Imaging biomarkers with an unlabeled drug
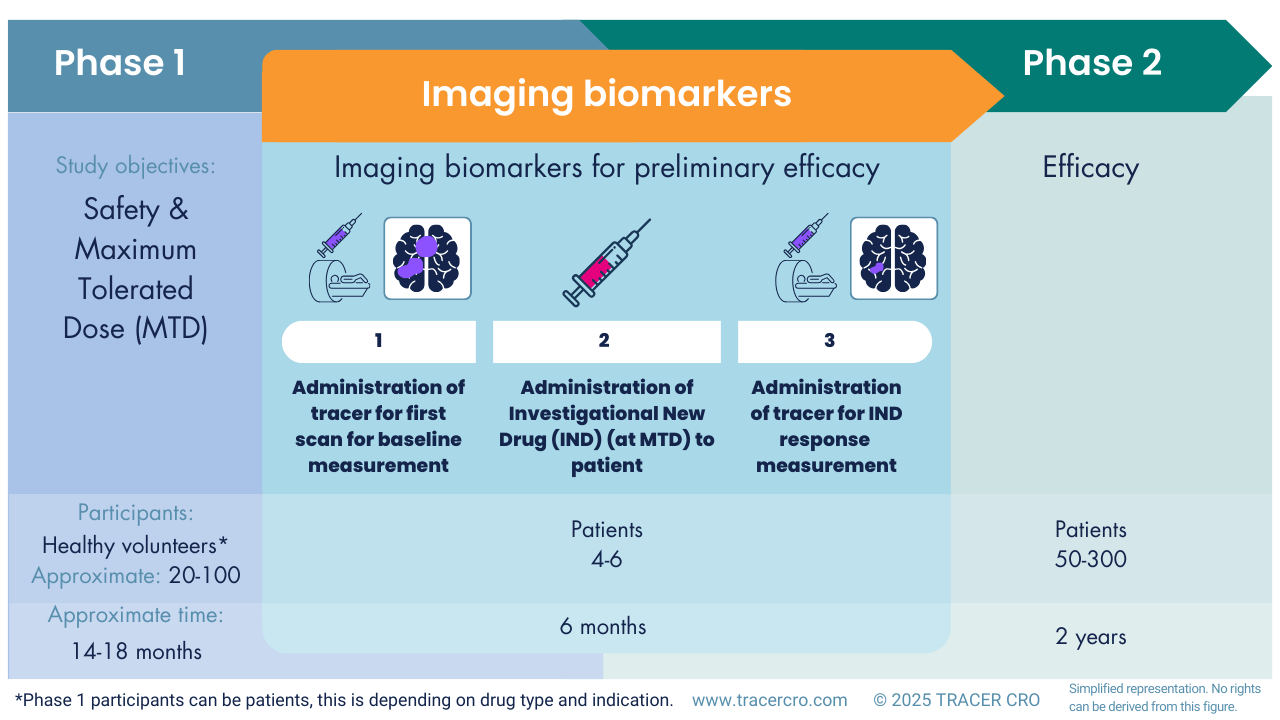
Labeling a drug is not always necessary or possible for nuclear imaging. In such cases a higher dose is often needed, but PET or SPECT imaging could still be of added value in early-stage clinical development. With separate administrations (tracer for the intended target, then unlabeled compound, and then the tracer again), it is possible to image target occupation to estimate treatment response. For example, in CNS drug development.
Radiotracer accumulation before and after IND treatment
An existing radiotracer for the same target can be used to evaluate preliminary efficacy. In this case, a baseline scan is acquired, after which patients will receive the IND in a standard Phase 1 safety and dose-finding study. After treatment, a second scan will be acquired to assess whether radiotracer accumulation is affected by IND treatment (as a surrogate marker for efficacy). Usually, these imaging markers show earlier changes than standard clinical and vital signs.
Competition of the IND with the radiotracer for the target
Another approach is to show competition of the IND with the radiotracer for the target. In a receptor occupancy study, an unlabeled drug and a PET tracer are administered [7]. If the IND is able to compete with the radiotracer for binding to the target, this is an indication that the IND binds the target. For this approach, the IND and radiotracer should bind to the same epitope of the target.
Other methods
PET, AMS, and LC-MS/MS are broadly used methods applicable to many drugs and research scenarios. Other methods are more limited in use but can be interesting to obtain a more detailed overall picture.
Fluorescence imaging
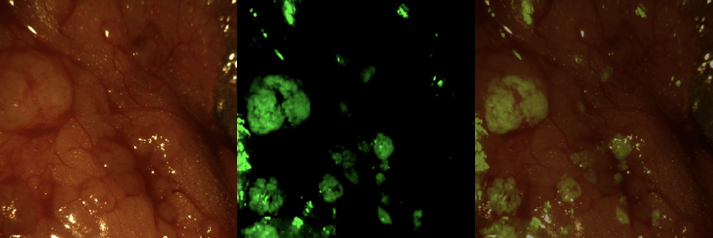
Fluorescent intra-operative imaging
Fluorescence imaging can be used to assess the presence of compounds at a cellular level. The maximum depth for measurements is 3 cm, however, based on various factors, 1-2 cm is a more conservative estimate. Fluorescence imaging is highly suitable for use during intraoperative procedures [15] or analyzing biopsies. [16]. The latter also demonstrating that Phase 0 is not limited to a microdose. The study included multiple cohorts, one using the microdose limit for biologics (30 nmol), but also groups that received higher doses (90 nmol and 171 nmol).
- The compound needs to be labeled with a fluorescent dye
- The fluorescent signal is suitable for semi-quantitative measurements
Raman
Raman uses a laser to measure light scattering, which enables the identification of molecules. This technique can be used to analyze the presence of a drug both topically and in samples.
- For Raman, the drug can be unlabeled
- The detection depth is limited by the reach of the laser used in Raman
- Compared to fluorescence, Raman doesn’t suffer from photobleaching, allowing longer periods of imaging.
- Strict Phase 0 studies using Raman are hard to find, but several studies underline the use of Raman as a valuable measuring tool [17, 18, 19, 20]
Raman with sub-therapeutic dose
Often, you need (sub)therapeutic concentrations on the skin to be able to measure this with Raman. However, by treating only a small area of skin, you can achieve local therapeutic concentrations on the skin, while the total dose in the entire body remains below microdosing levels. You will then need toxicity studies for topical administration to justify the concentrations in the clinical study. This is in addition to the single dose extended toxicity study i.v.
8. What data can be obtained from a Phase 0 study?
Depending on the methods used for measurements and whether participants are patients, healthy volunteers, or both, a Phase 0 study delivers data on:
- pharmacokinetics (limited to measurement of radioactivity concentrations)
- biodistribution
- on- and off-target binding
- time-activity curve
- dosimetry
- pharmacodynamics (to some extent)
- molecular pathway and mechanism
- disease biomarkers
- ADME and metabolites (limited to measurement of radioactivity concentrations)
- possible drug-drug interactions (DDI) (not in the standard Phase 0 study design)
9. How can Phase 0 data be used?
Phase 0 data is versatile in its use:
- Understand the biodistribution of your drug and on- and off-target accumulation by learning about binding, half-life, and residence time in organs and tissues.
- Study pharmacokinetics and biodistribution in your patient population.
- Compare different drug candidates to select the lead candidate based on human data rather than animal data.
- Study the biological pathway and mechanism in-human.
- Obtain time-activity curve data from emitted radioactivity.
- Limited ADME and metabolite data in humans, and to some extent food effect.
- Identify organs and tissues with a high uptake to predict potential adverse events.
- Compare delivery methods, predict absolute bioavailability (ABA), and calculate the therapeutic dose delivered. Calculated from a microdose by quantitative analysis of the PET and SPECT images (and dosimetry calculations for radiopharmaceuticals for targeted radionuclide therapy).
- Study biomarkers of a disease and (to some extent) the pharmacodynamics of your drug.
- Validate with human data the outcomes of non-clinical disease models and simulations (in-vitro and animal (model) data).
- Compare healthy volunteers (response, sensitivity) to patients.
- Compare different patient populations and disease subtypes.
- Compare the drug and disease target to existing therapies and the standard of care.
- Get an early estimate of drug-drug interactions (DDI).
Choose methods according to study objectives
This gives you a general idea of the many possibilities of Phase 0 studies. Keep in mind that the data is based on non-therapeutic dosing and on the method you choose. One method alone, PET, AMS, or LC-MS/MS, can’t deliver all the mentioned objectives. Depending on your study goals, you can decide on the method(s) to use. Contact TRACER to discuss your needs and get advice from imaging experts for your research design.
10. Is microdose data scalable to therapeutic levels?
The predictive value of microdose studies to therapeutic levels has been studied. A comparison of 57 microdose studies, 41 with an oral, and 16 with an i.v. administered drug, showed an adequate predictability of respectively 68% and 94% [21]. This is in line with earlier studies [22, 23, 24]. Most assessments evaluating the scalability of microdosing to therapeutic dosing use data from AMS studies, but PET shows a good correlation with AMS [12].
Extrapolation of microdose to therapeutic dose: factors of influence
Extrapolation between microdose and therapeutic dose may be influenced by metabolism (enzymes, transporters), saturation mechanisms, target expression, and target affinity. This applies especially for oral administration, where gastrointestinal processes are influential. At TRACER, we can assess if an investigational new drug is prone to nonlinearity and adjust the strategy accordingly.
11. Is Phase 0 a healthy volunteer or patient study?
A Phase 0 study may include patients or healthy volunteers. The use of a cohort of patients and healthy volunteers is also possible. Phase 0 can be conducted as a first-in-patient study, which can be used to study target engagement and disease-related factors. Within a patient population, multiple cohorts are also possible. For example, the study can have a basket trial design. Meaning multiple indications with different characteristics that share the same target can be included. Furthermore, even multiple compounds can be assessed, allowing for lead candidate selection [14]. There is considerable flexibility in the selection of cohorts for exploratory studies in humans.
12. How many participants are enrolled?
A Phase 0 study typically includes between 5-15 participants; the size depends on the study design. The number of participants may be higher when subjects are divided into several cohorts. For example, when including the lead and back-up compound, with a group of healthy volunteers and patients, or patients grouped in multiple indications.
13. How long does a Phase 0 study take?
The study itself takes only a few months to complete, depending on the type of patients and availability at the clinical site. From start to finish, the study can take 12 to 14 months. This includes conducting the required toxicity studies, labeling the drug for imaging or AMS (optional), and other steps in the areas of Chemistry, Manufacturing, and Controls (CMC), up to the completion of the Investigational Medicinal Product Dossier (IMPD), approved by a Qualified Person (QP) and Quality Assurance (QA). It also includes all regulatory matters, such as designing the study, writing the study documentation, submitting the application, and obtaining approval. And, finally, the selection of clinical sites, site preparation visits, staff training, trial implementation, image analysis, data validation, and writing of the trial report.
14. Can Phase 0 be used for all types of drugs?
Yes, Phase 0 is possible for chemical compounds and biologically derived products. Examples include small molecules, peptides, antibodies, antibody fragments, and nanoparticles. Even for Advanced Therapy Medicinal Products (ATMPs) such as cell therapies, Phase 0 is considered a possibility [25, 26], but additional regulatory requirements may apply. The possibilities are not limited to a specific type, as demonstrated by this Phase 0 study with gold nanoparticle cores conjugated with small interfering RNA oligonucleotides [27]. For biological drugs, there are some additional requirements. The maximum dose is 30 nmol, and ICH S6 [28] should be reviewed with regard to preclinical data.
15. What regulatory guidance applies to Phase 0 studies?
For exploratory trials in the US, the FDA provides the document Exploratory IND Studies [1]. The EMA lacks such guidance; its document, Position Paper on the non-clinical safety studies to support clinical trials with a single micro dose, is no longer available. For EU clinical trials with a microdose, EMA now refers to ICH M3 (section 7) [3]. Additional information can be found in the ICH guideline M3 (R2) – questions and answers [29] and The EMA Guideline Strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products – Scientific guideline [30]. For Japan, read Guidance for Conducting Microdose Clinical Studies (Notification No. 0603001 of the Evaluation and Licensing Division, PFSB dated June 3, 2008) [5].
16. Can other regulatory documents apply as well?
In addition to regulatory documents specific to Phase 0, others may apply. While those do not distinguish in requirements for Phase 0/microdose and other trials, nor for specific drug types, parts may apply depending on your study. We list a few important ones.
- ICH S6 (R1) Preclinical safety evaluation of biotechnology-derived pharmaceuticals – Scientific guideline [28]
- ICH S9 Non-clinical evaluation for anticancer pharmaceuticals – Scientific guideline [31]
- Microdose Radiopharmaceutical Diagnostic Drugs: Nonclinical Study Recommendations [32
- FDA Oncology Therapeutic Radiopharmaceuticals: Dosage Optimization During Clinical Development Draft Guidance for Industry August 2025 [33]
- Advanced therapy medicinal products (ATMPs) [34]
Regulatory documents depend on the region of your clinical trial
Depending on where your clinical trial takes place, there is specific guidance. All FDA clinical trial guidance documents can be found here. For European clinical trials, this EMA page is a good starting point. When planning clinical trials in the EU, keep in mind that EU member states (MS) have their own committees, like CCMO for clinical trials Netherlands.
When planning your clinical trial in a specific EU MS, take a look at “Overview part II requirements in a CT application per MS” [35]. All international, harmonized guidelines can be found on the ICH website.
Outdated regulations, still worth reading?
While the EMA position paper no longer applies, it’s still worth reading. It gives an interesting insight into the regulatory view on exploratory trials. The same applies to the “Guidance to the conduct of exploratory (phase 0) trials in Belgium”. The EMA document is no longer available for download, but when contacting TRACER, we can send it to you. The Belgium document is still available for download [36]. While these documents no longer apply, they give insight into study goals, design, and relevant drug product and nonclinical data from a regulator’s perspective.
Discuss what regulations apply to you
Contact TRACER ff you’re looking for an answer regarding regulations for your clinical trial. In our experience, the rules for Phase 0 are not clearly defined in regulatory documents. Often, less stringent rules apply when a microdose is used. A good example of this is the use of a Good Laboratory Practice (GLP)-grade drug substance in a Good Manufacturing Practice GMP-grade drug product. While nowhere clearly stated, labeling a GLP drug substance under GMP conditions makes it a GMP-grade product. This is a common practice for Phase 0, but not clearly covered in regulatory guidance.
Request a meeting
We are open to discussing which documents apply to you and which strategy you should follow. From a trial design perspective and from a regulatory perspective, TRACER can assist you. We are happy to help you obtain rapid approval with fewer requests for information from regulatory authorities.
17. Which toxicology studies are required in microdose clinical trial applications?
ICH M3 —summarized in our Exploratory trial approaches table—describes the required toxicology studies to include in your exploratory clinical trial application. They are also described in International Atomic Energy Agency (IAEA) Guidance for Preclinical Studies with Radiopharmaceuticals [37].
- with a single administration, you need a 14-day extended single-dose toxicity study with necropsy at days 2 and 14;
- with up to 5 doses, you need a 7-day repeated dose toxicity study;
- A single mammalian species is sufficient (with a control group); this is often a rodent study.
- In certain cases, and depending on the region, it is allowed to conduct non-clinical studies with the unlabeled compound and extrapolate findings to the radiolabeled substance.
- The NOAEL, or appropriate margin of safety should be established to use this in defining the maximum dose in the microdose study [5].
The required package of preclinical data depends on the type of compound and study design.
Toxicology studies for non-microdose exploratory trials
Exploratory trials commonly use a microdose, but higher doses are allowed. To use a higher dose, more preclinical data are required.
Most important deviations from the requirements of microdose studies:
- Rodent and non-rodent studies
- Genotoxicity is required
Need-to-know’s on preclinical studies
- All pivotal preclinical studies and safety, dosing, and toxicity studies need to be conducted under GLP guidance.
- The duration of the in-human study is restricted by the duration of your preclinical study.
- Regulators encourage combining different preclinical studies according to the 3R principle for Replacement, Reduction and Refinement in animal research.
18. What pharmacology data is needed for an exploratory clinical trial?
In addition to in vivo toxicological data, regulatory authorities also expect a certain degree of preclinical proof of concept to support human testing.
- In vivo / in vitro characterization of pharmacology.
- For radiolabeled drugs, this should also include radiation dosimetry and prove that labeling has no significant effect on pharmacology characterization.
- Non-microdose exploratory trials should include Ames assay, or an alternative if Ames is not suitable.
- For higher doses and repeated dosing, the results of a chromosomal damage test should be included.
19. Should preclinical data be GLP or non-GLP?
For basic research and drug discovery, GLP is not mandatory. Once potential drug candidates are prepared for clinical trials, for toxicology studies, regulators expect GLP studies or “in the spirit of GLP”. In general, toxicology studies always require GLP. For clinical trials, you’ll need GMP. Phase 0 imaging trials are performed while still in the preclinical stage, so developing the GMP-grade drug substance may come too soon. This issue is solved by labeling a GLP drug substance – which you already have at this stage – in a GMP facility. This is further explained in the next paragraph.
20. What are the CMC requirements for Phase 0 drug products?
Regarding Chemistry, Manufacturing and Control (CMC) you need GMP grade material, as is required for all in-human studies. In Phase 0, and only when the risk is considered low, several papers [38, 39, 40] describe the use of a GLP-grade drug substance (as a precursor) if the finalized, radiolabeled, drug product is manufactured under GMP. This also applies to the radionuclide that is used [41]. Requirements may vary depending on regional authority, drug type, dose, and indication. The GMP facility will perform quality control and analysis on starting materials to verify the characteristics and purity. The ICH and FDA microdose guidance do not clearly mention CMC requirements, but the Japanese guidance does [5].
21. Does radiolabeling a drug for clinical trials make it a radiopharmaceutical?
When the medicinal product is labeled with a radionuclide in clinical trials, do guidelines for radiopharmaceuticals apply? Yes, by labeling a drug with a radionuclide, additional guidelines apply. Which guidelines apply depends on the method and nuclide—PET (e.g. 11C , 13N , 15O , or 18F), SPECT (e.g. 123I , 99mTc , 111In), or 14C for AMS—and the dose. The FDA and ICH microdosing study guidance document don’t mention this specifically, but the Japanese guidance [5] does. Regional differences in the EU exist, as this paper describes [42], where the same 14C-labeled product was considered a radioactive formulation in the Netherlands, but not in the UK and Estonia. This difference can be explained by the dose, which is also mentioned in the Japanese guidance, where there is a limit to which 14C is subject to radiation regulations.
What regulatory guidance documents for radiopharmaceuticals are available?
Regulators provide guidance documents for the development of radiopharmaceuticals. They differentiate between diagnostic and therapeutic radiopharmaceuticals, approved and investigational new drugs, and whether the drug is intended as a radiopharmaceutical or only labeled for research purposes.
Authorities have specific guidance for radiopharmaceuticals
- FDA Oncology Therapeutic Radiopharmaceuticals: Nonclinical Studies and Labeling Recommendations Guidance for Industry [43]
- FDA Microdose Radiopharmaceutical Diagnostic Drugs: Nonclinical Study Recommendations [32]
- Guidance FDA Oversight of PET Drug Products Questions and Answers [44]
- Guidance for Industry and Researchers The Radioactive Drug Research Committee: Human Research Without An Investigational New Drug Application [45]
- Guidance for Conducting Microdose Clinical Studies (Notification No. 0603001 of the Evaluation and Licensing Division, PFSB dated June 3, 2008). [5] (No English document as an official translation available)
- Guideline for Clinical Evaluation of Diagnostic Radiopharmaceuticals [48] (English translation)
22. Are pediatric exploratory trials and trials with pregnant or lactating women possible?
Are Phase 0 exploratory trials possible in pediatric, pregnant, or lactating populations?
For clinical trials with children, international agreements are clear that:
- research should generally be conducted in adults first, especially regarding toxicity [49];
- pediatric studies may not pose more than the minimal risk;
- should benefit the individual participant;
- and provide data vital to the research of the disease or condition.
Although there are international and EU-harmonized regulatory guidelines, countries provide their own guidance. Keep in mind for radioactive compounds used in PET and AMS, radiation exposure limits need to be adjusted for pediatric populations [5].
FDA microdose studies: pediatric, pregnant and lactating women
While the EMA and ICH don’t explicitly exclude children and pregnant or breastfeeding women from exploratory trials, the FDA specifically states that it’s generally not permitted [1]. However, their 2012 presentation, “Ethical Considerations in Evaluating Non-Therapeutic Studies in Children” [50] mentions that pediatric studies are only considered when the risk is considered low and the study is expected to be of vital importance. The FDA perspective is further explained in this paper [51]. The use of sensitive technologies, such as AMS, also has an impact on the feasibility of pediatric studies [52, 42] and can contribute to a better understanding of age-related dosing [53].
EMA microdosing studies: pediatric, pregnant and lactating women
For clinical trials in the EU, drug developers should discuss their study with national authorities, which may provide their own guidance. For example, pediatric microdose studies have been conducted in the Netherlands, Estonia, and the UK [42]. Belgium however, specifically states “Exploratory CTA studies should not be performed in pediatric patients and pregnant or lactating women.” [36].
Microdose pediatric clinical trials in the Netherlands
For Clinical trials in the Netherlands, the ethics committee, CCMO, states that ‘No, unless’ principle applies for research with subjects under 16 [54]. As mentioned before, there are examples of pediatric microdosing studies in the Netherlands, as discussed here [55, 56]. To date, no exploratory microdosing studies have been conducted in pregnant or breastfeeding women, but the potential has been discussed [57].
23. What is cassette microdosing?
You may know “cassette dosing” (or N-in-One dosing) from animal studies as a higher-throughput screening [58], but did you know something similar is possible in-human? Cassette microdosing, as described by Burt, Tal et al. [59] means administering multiple drugs to the same subject at the same time. As long as you stay under the microdose definition, you can administer multiple drugs or compounds sequentially, almost at the same time. This way, you can measure the plasma concentration (bioavailability, PK) of different compounds, as this study shows [60]. This design also enables drug developers to study drug-drug interactions (DDI) [61]. Cassette microdosing is possible for i.v. administration as well as oral [62] and possibly other administration forms.
24. What is intra-target microdosing?
Intra-Target Microdosing (ITM) is a combination of microdosing and intra-target drug delivery. It differs from other types of microdosing by method of administration. The drug is locally administered; this can be done directly or via an incoming artery to the target. This leads to a locally higher, therapeutic level, but still with systemic sub-pharmacological exposure. ITM is similar to Intra-Arterial Microdosing (IAM) [63]. Drug developers can evaluate in-human target engagement, as these studies show [41, 64]. Fluorescence or PET imaging may be useful, but since the local dose delivered is higher, biomarkers may also be used to obtain data with ITM/IAM. The in-situ human data can be used to guide further decisions. Note: T. Burt holds the patent for Intra-Target Microdosing (ITM) [65].
25. What is a microdose cocktail?
A microdose cocktail differs from other types of microdosing studies. The method of microdose cocktail is used to study the drug-drug interaction (DDI) of various drugs administered simultaneously or, in some cases, sequentially. [66]. These type of studies can be conducted with healthy volunteers, patients or both [67, 68]. Microdose cocktail should not be confused with cassette microdosing, in which multiple related formulations of the investigational compound are administered. At the moment, studies with multiple compounds are not using imaging. PET imaging is suitable for imaging one compound at the time, although developments in multi-isotope imaging are made. SPECT does allow multi-isotope imaging, which can be used in animal studies.
Conclusion
We’ve answered 25 questions on Phase 0 exploratory microdosing trials and referenced relevant guidance documents, review papers and studies that we list below. If you have any other questions, do not hesitate to contact us. Phase 0 offers a great opportunity for drug developers. A quick search on Clinicaltrials.gov reveals that over 5,000 early Phase 1 clinical trials have been conducted since the beginning of this program. There’s a large deal of variation in study contents, showing there is interest from the industry and flexibility from regulators in what study types to approve. At TRACER we are happy to discuss your ideas and provide input from our expertise. Feel free to contact us.
Abbreviations
| ABA | Absolute Bioavailability |
| ADME | Absorption, Distribution, Metabolism, and Excretion |
| AMS | Accelerated Mass Spectrometry |
| ATMP | Advanced Therapy Medicinal Product |
| AUC | Area Under the Curve |
| BD | Biodistribution |
| CMC | Chemistry, Manufacturing, and Controls |
| CRO | Contract Research Organization |
| DDI | Drug-Drug Interaction |
| FDA | U.S. Food And Drug Administration |
| GLP | Good Laboratory Practice |
| GMP | Good Manufacturing Practice |
| ICH | The International Council For Harmonisation Of Technical Requirements For Pharmaceuticals For Human Use |
| IAEA | International Atomic Energy Agency |
| IAM | Intra-Arterial Microdosing |
| IMPD | Investigational Medicinal Product Dossier |
| IND | Investigational New Drug |
| ITM | Intra-Target Microdosing |
| i.v. | Intravenous |
| LC-MS/MS | liquid Chromatography-Tandem Mass Spectrometry |
| NOAEL | No Observed Adverse Effect Level |
| nmole | Nanomole |
| PAD | Pharmacologically Active Dose |
| PET | Positron Emission Tomography |
| PK | PharmacoKinetics |
| QA | Quality Assurance |
| QP | Qualified Person |
| SPECT | Single-Photon Emission Computed Tomography |
| μg | Microgram |
Citations
https://english.ccmo.nl/investigators/additional-requirements-for-certain-types-of-research/research-with-participants-under-the-age-of-16-years-children
Although this article has been composed with great care and attention, we cannot guarantee its accuracy. If you have any suggestions or additions to this article, please email info@tracercro.com.
No rights can be derived from this publication. This blog does not make claim or promote ownership to any intellectual property, study information, or copyrighted terms wherein.